



Anvisa aprova o primeiro produto de terapia gênica no país, indicado para o tratamento da distrofia hereditária da retina.
A Anvisa publicou, no Diário Oficial da União (D.O.U.) desta quinta-feira (6/8), o registro do primeiro produto de terapia gênica no Brasil, indicado para o tratamento de doença rara. Trata-se do Luxturna® (voretigene neparvoveque), um tipo especial de medicamento de terapia avançada, denominado produto de terapia gênica, produzido pela Novartis Biociências S.A.
O produto é utilizado em crianças acima de 12 meses e em adultos com perda de visão, para o tratamento da distrofia hereditária da retina, causada pela mutação do gene humano RPE65. Dentre as distrofias hereditárias da retina estão a amaurose congênita de Leber e a retinose pigmentar. A mutação do gene RPE65 causa a ruptura gradual das células localizadas na parte de trás do olho que formam a retina, causando perda gradual da visão, geralmente na infância ou na adolescência, que progride para cegueira. Até o momento, não havia alternativa terapêutica para a doença.
Elaborado por engenharia genética, o produto é composto por um vírus onde se inseriu cópia do gene humano RPE65, responsável pela produção de uma enzima necessária para o funcionamento normal da retina. Essa enzima permite um melhor funcionamento das células da retina, diminuindo o progresso da doença. O vírus utilizado na fabricação deste produto não causa doença em humanos.
É importante esclarecer que se trata de um produto de uso hospitalar e sob supervisão médica especializada, administrado via injeção sub-retiniana, e que devem ser adotadas precauções, como a obrigatoriedade de o paciente passar por testes capazes de comprovar que sua perda de visão foi provocada por mutações no gene RPE65, sobre o qual a substância ativa do Luxturna atua, além de outros critérios laboratoriais e clínicos que devem ser avaliados pelo médico.
Marco regulatório
A Agência aprovou recentemente o marco regulatório para o registro de produtos de terapia avançada no Brasil, a Resolução da Diretoria Colegiada (RDC) 338/2020. Essa norma criou um ambiente regulatório seguro para a aprovação de produtos dessa natureza no país.
O processo de registro de um produto de terapia avançada, por exemplo, de terapia gênica, envolve a análise da comprovação de segurança por meio de dados robustos de experimentos pré-clínicos, bem como de segurança e eficácia por meio de resultados de estudos clínicos capazes de evidenciar os benefícios ao paciente em determinada dose e posologia terapêutica.
Faz-se necessária ainda a comprovação de produção com requisitos de qualidade e boas práticas de fabricação, de estudos de mecanismos de estabilidade e distribuição do produto, além de cuidados especiais ao paciente, previsão de eventos adversos possíveis, orientações ao profissional da saúde que exercerá o cuidado ao paciente, definição de monitoramento e gerenciamento de risco pós-uso, dentre outras análises pertinentes para garantir que o produto e o processo a serem registrados na Agência estejam adequados ao uso terapêutico, principalmente neste caso, que envolve também pacientes pediátricos.
Considerando as especificidades dos produtos de terapia gênica e as suas características inovadoras, a Anvisa firmou um termo de compromisso com a empresa detentora do registro no país, no sentido de assegurar a realização de estudos complementares com pacientes brasileiros, de forma a acompanhar o perfil de segurança e de eficácia do produto em longo prazo. As condições impostas ao detentor de registro serão informadas ao público, juntamente com os prazos e datas para sua execução, por meio do portal da Agência.
Por se tratar de um organismo geneticamente modificado, o produto ainda foi avaliado em relação à sua biossegurança, pela Comissão Nacional Técnica de Biossegurança (CTNBio), que também se manifestou favorável à sua aprovação.
Fluxo
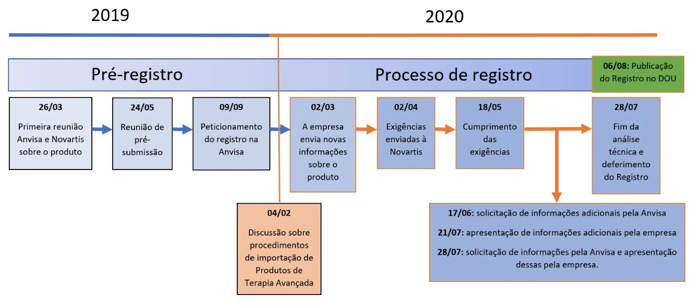
Foram 232 dias úteis até a aprovação da Anvisa, considerando desde a submissão dos documentos, os prazos de análise da equipe da Agência e as respostas ao cumprimento das exigências por parte da empresa. Ocorreram ainda reuniões e comunicações técnicas entre a Anvisa e representantes nacionais e internacionais da empresa.
Tendo em vista a complexidade do produto, os prazos aplicados na Anvisa foram compatíveis com as principais agências reguladoras do mundo, como a americana Food and Drugs Administration (FDA) e a europeia European Medicine Agency (EMA), que levaram, respectivamente, 372 e 331 dias úteis.
Confira a linha do tempo até a publicação do registro:
Com informações do site da ANVISA (06/08/2020)